CIENCIAS DE LA SALUD-Artículo Científico
MIGRATION OF Leishmania (Viannia) panamensis AND ITS PERSISTENCE IN HEALTHY SKIN OF HAMSTER
MIGRACIÓN DE Leishmania panamensis Y SU PERSISTENCIA EN PIEL SANA DE HAMSTER
Lina M. Carrillo-Bonilla1, Andrés Montoya2, Natalia Arbeláez3, Horacio Cadena4; Jaime Ramírez5, Sara M. Robledo6
1 MV. M.Sc., Profesora asociada Facultad de Ciencias Agrarias, Universidad de Antioquia, Medellín, Colombia, lina.carrillo@udea.edu.co; Autor de correspondecia
2 MyB., Estudiante de Maestria en Biologia, Grupo de investigación PECET, Universidad de Antioquia, Medellín, Colombia, andres19@gmail.com
3 MV., PECET, Universidad de Antioquia, Medellín, Colombia,natyac182@gmail.com
4Biolg. M.Sc., PECET, Universidad de Antioquia, Medellín, Colombia: horacio.cadena@udea.edu.co
5 Biolg. Esp. Instituto Nacional de salud -INS-. jramireza@ins.gov.co
6 MyB. PhD., PECET, Universidad de Antioquia, sara. robldo@udea.edu.co Calle 70 No. 52-21, Medellín, Colombia
Rev. U.D.C.A Act. & Div. Cient. 17(2): 341-350, Julio-Diciembre, 2014
SUMMARY
Leishmaniasis are endemic in >98 countries with 14 million people infected. Leishmania survives in mammal hosts even after treatment but the capacity to persist in healthy skin and infect sandflies from these sites is poorly understood. To demonstrate migration of Leishmania (V.) panamensis into healty skin, persistence in these sites during chronic stages of infection and infectivity of persistant parasites to Lutzomyia longipalpis 10 hamsters were inoculated intradermally in the hind footpad, with 2x107 L. (V.) panamensis promastigotes. Hamsters were sacrificed at 13 (acute phase) and 53 (chronic phase) weeks post-infection. Biopsies from lesion, healty skin and othert tissues were screened by Hsp70 polymerase chain reaction (PCR) and DNA polymerase qPCR. Transmission of parasites to sandflies was determined by xenodiagnosis. Positivity was observed in all samples of acute infection group except the contralateral ear and chronic infection group, although the percentage of positivity in chronic infection was lower than that for acute infection. Two animals of the acute infection group and one from the chronic infection group were infective to sand flies after feeding over the lesion site. No infections were found in sandflies fed from healthy skin. Dissemination and persistence of L. (V.) panamensis was reported for first time and persisntace is confirmed for a period of 53 weeks. However, parasite load in tissues different from the inoculation site was lower and transmission to the sand fly vectors from these sites was not observed. Hsp70 PCR and qPCR are useful techniques for determining infection status of vertebrate hosts.
Key words: Parasite migration, xenodiagnosis, Hsp70 PCR, real-time DNA polymerase
RESUMEN
Las leishmaniasis son endémicas en >98 países con 14 millones de infectados. Leishmania sobrevive en los mamíferos aún después del tratamiento pero se desconoce su capacidad de persistir en piel sana e infectar vectores. Para demostrar la migración de Leishmania (Viannia) panamensis a piel sana, su persistencia en estos sitios durante estados crónicos de infección y la infectividad de los parásitos persistentes para Lutzomyia longipalpis, se inocularon 10 hámsters intradérmicamente en pata con 2x107promastigotes de L. (V.) panamensis. Los hámsters se sacrificaron a las 13 (fase aguda) y 53 (fase crónica) semanas post-infección. Biopsias de lesión, piel sana y otros tejidos se analizaron por reacción en cadena de la polimerasa (PCR) con el gen Hsp70 y qPCR con el gen DNA polymerase. La transmisión de los parásitos a Lu. longipalpis se determinó mediante xenodiagnóstico. Todas las muestras de fase aguda (excepto oreja contralateral) y todas las muestras de fase crónica fueron positivas, aunque la positividad en fase crónica fue menor que en fase aguda. Dos animales de fase aguda y un animal de fase crónica fueron infecciosos al vector, después de alimentarse sobre la lesión. No hubo ningún insecto positivo en los alimentados sobre piel sana. Se reporta por primera vez la diseminación y persistencia de L. (V) panamensis, y se confirma la persistencia en un periodo de 53 semanas. Sin embargo la carga parasitaria en tejidos diferentes al sitio de inóculo fue baja y no se detectó su transmisión al vector. La PCR con Hsp70 y qPCR con DNA polymerase son técnicas útiles en la determinación del estatus infeccioso en los hospederos vertebrados.
Palabras clave: Migración parasitaria, xenodiagnóstico, PCR Hsp70, ADN PCR en tiempo real
INTRODUCTION
Leishmaniasis is a group of diseases endemic in 98 countries of the tropical and subtropical geographical regions. The disease varies in clinical presentation, including cutaneous (CL), mucosal (MCL) and visceral leishmaniasis (VL). Annually, two million new cases are recorded and 350 million people are estimated at risk. The disease is ranked ninth in terms of global burden of all infectious and parasitic diseases, and is considered the most neglected in this category (Alvar et al. 2012).
Evolution of Leishmania in the vertebrate host tissues after infection is fundamental in the search for new strategies for both treatment and control of leishmaniasis. Leishmania species are able to thrive in their vertebrate hosts, being immunologically competent, they persist even after clinical resolution of the disease or in animals infected without symptoms (Bogdan et al. 1996; Mendonça et al. 2004). From the point of the view of the vertebrate host, parasite persistence is important in maintaining immunity through constant antigen presentation to the immune system. However, from the point of view of the parasite persistence is important to ensure its own transmission, and therefore, maintaining its life cycle.
L. (V.) panamensis is one of the most prevalent species in Colombia and other countries in Latin America causing CL but also MCL (González & Vélez, 2006). However, little is known about the persistence of L. (V.) panamensis in healthy skin of infected vertebrate hosts (reservoirs) that would facilitate their ingestion during the vector's blood meal (Roque et al. 2014).
The golden hamster Mesocricetus auratus is widely considered an adequate animal model for experimental CL since it is highly susceptible to infection by all Leishmania species and develops similar lesions to those observed in humans. These animals also develop chronic non-healing skin lesions with a small percentage of the animals developing late dissemination to other sites (Travi et al. 2002; Hommel et al. 1995; Osorio et al. 2003; Robledo et al. 2012). Some studies demonstrated L. (V.) pamanensis migration via lymphatic after observed lesion in other body parts (Martínez et al. 1991), but never analyzed their presence in healthy skin. The dispersion of Leishmania through the lymphatic system is supported by the frequent presence of lymphadenopathy and the ability to isolate parasites from lymph nodes draining the site of the lesion (Barral et al. 1995; Santos et al. 2014).
To achieve a better understanding of Leishmania persistence in the vertebrate host and its implications in the transmission of leishmaniasis, this study aimed to evaluate the ability of L. (V.) panamensis to migrate and survive in the healthy skin of hamsters experimentally infected in the hind footpad.
Furthermore, it was determined if parasite present in places different from the inoculation site were infective for the sand fly vector.
MATERIAL AND METHODS
Ethical consideration: This protocol was approved by the Animal Experimentation Ethics Committee of the University of Antioquia, Act 56, issued on September 15, 2009, and these followed all of the international standards of animal welfare.
Animals: Ten adult males and females of the inbred golden hamster (6-8 weeks old, weighing 100-120g) were used. Hamsters were kept in the animal facilities at the University of Antioquia, Medellin, Colombia under temperature and moisture-controlled accommodation, fed with standard rodent dried food and provided with sterile water ad libitum.
Parasites culture: L. (V.) panamensis promastigotes (MHOM/ CO/87/UA/UA140) in stationary growth phase were cultured in biphasic Novy-MacNeal- Nicolle (NNN) medium (Walton et al. 1977). Promastigotes were washed in phosphate-buffered saline, 0.15 M, pH7.2 (PBS) and adjusted to a concentration of 2X107 parasites in 100µL.
Animal infection: Prior to experimental infection, hamsters were anesthetized intraperitoneally with a 9:1 mix of Ketamine (50mg/kg) and Xilacine (20mg/kg) in a volume of 260-300µL. Two groups of five animals each were formed. Group 1 (acute infection) included 3 male and two female hamsters having 13 weeks post inoculation while Group 2 (chronic infection) included 3 male and two female hamsters with 53 weeks post inoculation. A group of three animals were inoculated with PBS and used as a negative control.
Animals were inoculated intradermally in the right hind footpad; then, they were monitored every 7 days for up to four - six weeks after inoculation for appearance of lesions. During the monitoring, the type of lesion was described and the induration and ulcer area were measured with a digital caliper. The presence of lesions in different regions to the inoculation site and the appearance of relapses of the lesion were also described and registered. Every two weeks, animals were weighted and lesions were pictured. Animals were observed daily to monitor physical appearance and behavior.
Tissue samples: Once the follow up was ended hamsters from both groups were sacrificed and parasite presence, load and distribution in the different tissues were assessed. Animals were euthanized by inhalation of CO2 The tissues were sampled as follows: lesion site (right hind footpad), skin from three (adjacent site) and six centimeters (distant site) away from inoculation site, popliteal draining lymph node, spleen, liver, and both ears (separately). Tissues were removed by scissors or scalpel, placed in petri dishes containing PBS with antibiotics, and stored at 4°C overnight. Samples were then carefully homogenized in 1.5mL vials with PBS. Aliquots were stored at -20°C until DNA extraction.
Detection of L. (V.) panamensis Hsp70 DNA: Tissue samples were weighted and DNA was extracted and purified using the DNeasy® blood and tissue kit (Qiagen, Boston, US) following manufacturer's instructions. The DNA was dried at room temperature, resuspended in 200µL of nuclease free water, quantified using NanoDrop 1000 (Thermo Scientific®, New Hampshire, USA) with 260nm of absorbance and stored at -20°C until further use. The PCR was performed using the oligonucleotide primers from hsp70 234pb 5'- GGA CGA GAT CGA GCG CAT GGT-3' and 5'-TCC TTC GAC GCC TCC TGG TTG-3', reported to specifically amplify the entire 234- bp Leishmania Hsp70 gene region (Graça et al. 2012). The PCR amplifications were performed in a final volume of 25µL containing 2µL of DNA, 20 pmol of each primer, 250µM dNTPs, 1.5mM MgCl2 and 1 U Taq polymerase (#EP0406, Thermo Scientific). The PCR conditions were: one cycle at 94°C for 5 min followed by 32 cycles of 94°C for 30 sec, 63°C for 1 min and 72°C for 1 min and a final extension at 71°C for 9 min. The end-point PCR products from the different assays were visualized on an ethidium bromidestained 2% (w/v) agarose gel. The amount of DNA per mg of tissue was determined. Total DNA from L. (V) panamensis grown in Schneider's Drosophila medium at 26°C was used as positive control.
Quantification of L. (V. ) panamensis DNA polymerase: Quantification of L. (V.) panamensis was based on amplification of the DNA polymerase single copy gene that is present in different species of Leishmania with a quantitative real time PCR (qPCR). Initially, a 600 bp fragment of DNA extracted from L. (V.) panamensis promastigotes was amplified by conventional PCR using the forward 5'- CTGCGCT- GTGTTATCCACAGC-3' and reverse 5'-GCTGTTTGCCGT- CAACTTCAG-3' primers in a thermocycler (BioRad) with the following cycles: 94°C for 10 min (denaturing) followed by 45 cycles of 94°C for 1 min (denaturing), 63°C for 1 min (annealing) and 72°C for 45 sec (extension) and a final extension at 72°C for 45 sec. The amplification product was v rified on a 1% (w/v) agarose gel. The fragment was excised and purified using the QIAquick Gel Extraction Kit according to the manufacturer's instructions. The purified product was ligated to InsTAclone pTZ57R / T vector. E. coli DH5a cells (Invitrogen, US) were transformed with the construct obtained above as follows: 50mL of competent cells maintained in freezing buffer (10mM MOPS, 10 mMRbCl, 75 mM CaCl2, 15% glycerol) were mixed with 5mLof the ligation product and incubated for 30 minutes on ice; then, cells were transferred for 90 seconds at 42°C and immediately returned to ice for 5 min. One mL of liquid Luria Bertani medium (LB medium) without antibiotics was added and, the mixture was incubated at 37°C for 1 hour. After this, transformed cells were centrifuged at 14,000rpm for 2 min and the superna tant was decanted. The cell button was resuspended in LBagar medium supplemented with ampicillin (100mg/mL) and incubated for 16 hours at 37°C. Ten colonies were randomly picked and checked for the presence of the pTZ57R/T- DNA polyerase construct by plasmid DNA extraction - Miniprep as follows: each colony was inoculated into 5mL of LB medium containing 100mg/mL ampicillin for 16 hours at 37°C under constant agitation (250rpm); plasmids were then recovered using the QIAprep Spin Miniprep Kit (Qiagen) according to manufacturer's recommendations. Once purified, the presence of the insert in the plasmid was verified by PCR and the plasmid DNA was sequenced. qPCR was set up using the forward 5'- TTCCGCTTGCCATCCTCCTC-3' and reverse 5'-TGAGCGCATCGAGTACCTCCTG-3' primers which amplify a 120 bp fragment within the 600bp sequence initially cloned. The Tm of the primers was set at 60°C. The presence of the fragment in the plasmid DNA, and L. (V.) brazilensis, L. (V.) panamensis, and L. (L.) infantum was verified by conventional PCR. DNA from uninfected hamsters was used as negative control.
The number of copies per plasmid was determined based on the size of the cloned fragment and the size of the insert. A standard curve ranging from 1 to 1 million parasites in log base 10 increases was established. The QuantiFast SYBR Green qPCR kit (Qiagen) was used for qPCR. The PCR amplifications were performed in a Smart Cycler II using a final volume of 25µL containing 1µL of DNA, 12.5 12.5µL of the mix, 100nM of each primer and nuclease free water. The PCR assays used the following amplification cycle: 95°C for 5 min followed by 40 cycles and 15 sec at 95°C, 20 sec at 60°C and 20 sec at 72°C, ending with a melting curve between 60°C and 90°C. The amplification efficiency of each was measured using the PCR program LinRegPCR versión 2013.1 (Ruijter et al. 2009).
Xenodiagnosis: Lu. longipalpis females F1 from the laboratory colony of Colombian National Health Institute (Bogotá, Colombia) were maintained at 26°C with a 14/10 LD photoperiod until use. One hundred twenty two sandflies were placed in round plastic boxes (10cm diameter X 5cm height) with an open side covered by a fine-mesh nylon screen. Sandflies contained in each box, were fed on inoculation sites, hind footpad, (Box 1) and contralateral hind footpad (Box 2), apparently healthy skin. Hamsters anaesthetized with intramuscular ketamin/xylaz (50 and 20mg/kg, respectively) were placed in the cage with Lu. longipalpis for 30 min. Subsequently, sandflies were transferred to holding cages that were kept at 25 - 28°C and 90% of relative air humidity. Cotton wool soaked in 20% sucrose was placed on the top of the cage to provide a continuous source of water and sugar.
On the sixth day after the blood meal, sand fly midguts were drawn out in a drop of sterile PBS and examined under the microscope for the presence of promastigotes.
Data analysis and statistical methods: Quantitative results are expressed by interpolation with a standard curve included in each qPCR run and consisting of a series of tenfold concentration of L. (V.) panamensis DNA polymerase: 1 copy/ ìl, 10 copies/ìl, 100 copies/ìl, 1,000 copies/ìl, 10,000 copies/ìl, 100.000/il and 1,000,000 copies/ìl. This curve was compared with a standard curve using DNA from a known parasite number: 5, 25, 125, 625, 3125, 15125, 78125 and 390625 promastigotes of L. (V.) panamensis. The detection threshold for the qPCR was set to >30 arbitrary units of fluorescence. For each of the samples and the dilutions of the standard curve the CT value corresponded to the cycle at which this fluorescence threshold is exceeded. This CT value was used to determine the parasite load in the sample using a linear regression built with the CT value of standard curve. Finally, the melting curve confirmed the amplification of the expected fragment with the separation of double-stranded DNA at 85°C in the standard curve, the DNA of the parasite and samples.
The PCR and qPCR results were analyzed according to the type of sample and the study group with the SPSS 18.0 software to identify associations between different categories applying the chi2 test. Cells with expected values less than 5, used Fisher's correction test. P value < 0.05 was considered significant.
RESULTS AND DISCUSSION
Significant changes in the body weight of hamsters were not detected during the study. The average weight was 136.4g SD+/- 2.48 and 134.7g SD +/- 3.04 in the chronic group. The average weight in the control group was 149.1g SD +/- 1.8 and 138.8g SD +/- 2.1 for the acute group and the chronic group respectively. Significant differences were not identified.
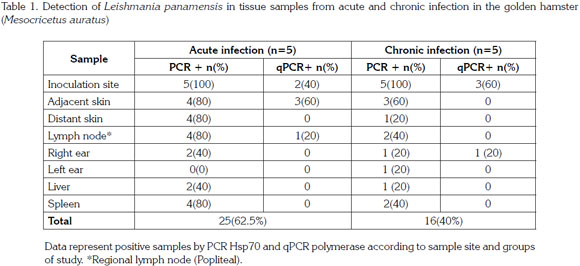
Lesions in tissues different from the site of inoculation were not observed. Animals in the acute stage of infection showed induration without ulcer with an average size of 2.5mm2, and all animals from the chronic infection group had induration and ulceration with an average size of 2.5mm2 and 3.5mm2, respectively. Clinical symptoms, induration and ulceration, only developed where the inoculation occurred; on other cutaneous tissues where parasites were detected, no clinical symptoms were observed. This finding could be due to the inoculum size used in the present work. In previous studies where higher inocula were used, parasite migration and lesion development was evidenced in distant tissues to the site of inoculum, such as the contralateral hind footpad (unpublished). During the acute and chronic phases of infection, 13 and 53 weeks post inoculation, respectively, parasites were observed in all tissues sampled with the exception of the contralateral ear that was negative in animals from acute phase (Table 1).
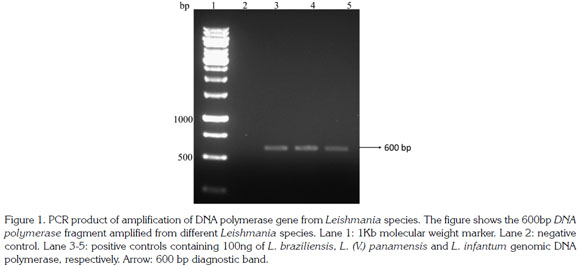
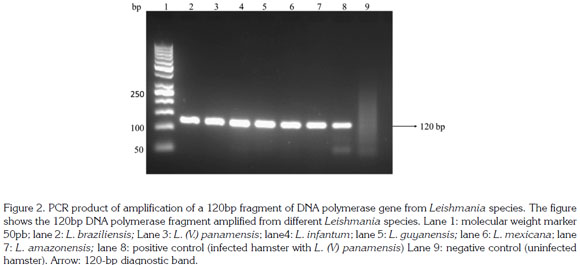
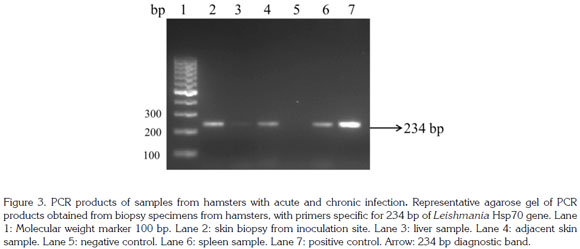
A 600pb fragment was amplified, purified and cloned (Figure 1). The cloning construct was quantified in NanoDrop 1000 (Thermo Scientific) and a standard curve was built. The qPCR amplified fragment was 120 bp from the initial fragment of 600 bp and was amplified in six Leishmania species: L. (V.) braziliensis, L. (V.) panamensis, L. (L.) infantum, L. (V.) guyanensis, L. (L.) mexicana and L. (L.) amazonensis) (Figure 2). The genomic DNA of uninfected hamsters was negative and therefore used as control. In samples from hamsters, amplification was achieved from 10 gene copies and the efficiency determined by LingReg was > 1.85 (Figure 3).
Results obtained by qPCR using both the standard curve absolute quantification method and a relative quantification method to estimate the number of Leishmania in experimentally infected hamsters are shown in figure 4. The parasite load in the inoculum site, determined by qPCR, was similar for both groups with values of 308 SD +/- 7.2 and 285 SD +/- 6.3 parasites per mg of tissue in the acute and chronic infection, respectively. Parasite load in adjacent skin in acute infection was of 280 SD +/- 15.2 parasites per mg of tissue while in the chronic infection group the parasite load only was determined for samples from the ipsilateral ear with values of 292 per mg of tissue. In the popliteal lymph node of both groups parasite load was of 288 parasites per mg of tissue. There were no significant statistical differences in parasite load between any of the experimental groups.
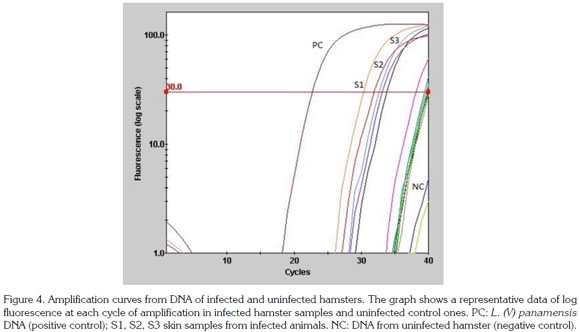
Although there are not significant differences, higher parasite load was detected in tissues from the inoculation site of hamsters with acute infection compared to the chronic group. This observation could be explained by the elimination that many parasites may suffer after the challenge of the immune system occurs. Additionally, the parasite needs to remain in small numbers inside the host so it is not easily recognized and attacked by the immune system. Another interesting finding is the presence of parasites in the ears. Ear tropism could represent a mechanism of the parasite to persist in the host and thereby ensure their intake by the vector and the perpetuation of their life cycle in the nature and transmission. The ear is made up of highly vascularized tissue, and covered with fur, being easily accessed to the sandflies for a blood meal.
The presence of parasites in skin away from the inoculation site highlights the importance of using ears in xenodiagno sis. However, the implications of the parasite's presence in the regional draining lymph node are twofold. Martínez et al. (1991; 2000) found that decreased frequency of isolation from the inoculation site and draining lymph nodes over time was accompanied by increased frequency of isolation from distant lymph nodes when the hamster was inoculated with L. (V.) panamiensis. From the point of view of the host, it is a challenge of this stimulus to the immune system, while for the parasite it could represent a temporary site until it reaches different tissues. Although it is necessary to determine whether the parasites detected are dead or alive, the parasite load detected by qPCR suggests that parasites could be alive. Studies on parasite viability in extralesional sites in humans have shown parasite survival through the measurement of RNA, but their infectiousness to vectors has not been proven (Romero et al. 2010).
Parasite burden is traditionally estimated by quantification of parasites in tissues using the limiting dilution technique (Titus et al. 1985), a poor sensitivity time consuming method. Thus, it was necessary to develop a more sensitive and easier method to estimate parasites in different tissues. In the last decade qPCR for estimating Leishmania infection in vertebrate hosts have been developed, based upon genes such as albumin, DNA pol, kDNA, SSU rDNA, ITS gen, among others (Davoust et al. 2013; de Paiva et al. 2013; Toz et al. 2013). Most of them show high sensitivity, accuracy, and reproduc ibility, demonstrating their utility to quantify amastigotes in tissues from naturally or experimentally infected hosts. The qPCR is a highly specific technique for the amplification of DNA from different species of Leishmania. Additionally, the technique is useful to determine the number of parasites per mg of tissue or blood in a quantitative and fast way (de Ruiter et al. 2014), unlike limiting dilution, although cheaper, may take up to a month in culture and has high risk of contami nation Other techniques such as the Giemsa stained smear do not determine the exact amount of parasites and requires highly trained personnel to read the samples. The qPCR is an easier, faster and accurate technique to quantify the parasite load.
The PCR technique based on amplification of the Hsp70 gene PCR allowed the detection of the parasite in various tissues different to where inoculation occurred. The gene encoding the cytoplasmic heat shock protein 70 kDa (hsp70) in the Leishmania parasite (Garcia et al. 2004) is highly conserved in many organisms and has maintained its function despite strong selective pressures (Folgueira & Requena, 2007; Fraga et al. 2010). Amplification of this gene has proven useful in the identification of several New World Leishmania species such as L. (V.) panamensis, L. (V.) braziliensis, L. (V.) guyanensis, L. (L.) amazonensis, L. (L.) infantum and L. (V.) lainsoni, demonstrating its applicability to the study of clinical samples from patients and mammals (Garcia et al. 2007, Montalvo et al. 2008; 2010). Moreover, the sequence of primers used in this work has proven to be more sensitive (Graça et al. 2012). Additionally, qPCR technique based on amplification of DNA polymerase gene was standardized. This method not only permits quantification of L. (V.) panamensis amplification but also allows optimization of the experimental model by measuring the parasite load in experimental infections with different Leishmania species. In this study, a greater sensibility of the hsp70 PCR was demon strated with respect to the DNA pol qPCR. This finding could be explained because the Hsp70 is a multicopy gene while the DNA polymerase is a single copy gene. The sensitivity of a qPCR based on amplification of Hsp70 gene remains to be determined. Finally, the long-term persistence of L. (V.) panamensis in healthy skin and other tissues was demonstrated in experimentally infected hamsters.
All surviving sandflies were dissected to determine the infection rates, which was 3/269 in acute group and 3/320 in chronic group. Two of the hamster from the acute infection group and one hamster from the chronic infection group were infective to sandflies after feeding on the lesion. No infections were found in the sandflies fed from contralateral hind footpad (healthy skin). Of the total of sandflies used in xenodiagnoses 47% on average were fed and 4.28% were positive for Leishmania promastigotes. These promastigotes was identified as Leishmania spp. by PCR. The ability of these parasites to infect Lu. longipalpis was also demon strated for parasites taken from the inoculation site, although in the present work xenodiagnosis was confirmed as poorly sensitive. In the same way any xenodiagnoses was positive in patient with active lesson of CL and just 5 of 57 patients were positive in xenodiagnoses from the lesion (Vergel et al. 2006). This may be due to the method used to detect infection in the sandflies, as dissection requires high expertise not only to dissect the specimen but also to find parasites. It would be interesting to assess infection in the sandflies using PCR techniques.
Previous studies on persistence of L. (L.) major in the mouse model with subcutaneous injection in the ear demonstrated the presence of parasites not only in the inoculation site but also in draining lymph nodes, blood, blood filtering tissues, and distant cutaneous sites, such as the contralateral ear and tail skin. In all other cutaneous tissue reached by parasites no clinical symptoms were observed (Nicolas et al. 2000). Similar results were observed in the present work.
A pilot test with four hamsters was carried out inoculating a higher dose of parasites in the snout. Animals were humanely sacrificed after 13 weeks (acute infection phase) and samples from inoculum site, adjacent and distant skin, liver, spleen and cervical lymph node were taken and processed in the same way described above. Positive results were obtained from samples from inoculum site and liver from all animals, samples from the adjacent skin and cervical lymph node from three animals and, samples from distant skin and spleen from two hamsters. However, it is necessary to evaluate the behavior of parasites inoculated into the snout during chronic phase of infection.
In summary, migration and persistence of L. (V.) panamensis was confirmed in hamsters experimentally infected. A decrease in the parasitic load during chronic phase was also confirmed. The utility of Hsp70 PCR and DNA polymerase qPCR techniques for determining the infection status of potential reservoirs and even for performing diagnosis of infection is also demonstrated. Because Hsp70 PCR was more sensitive than DNA polymerase qPCR, the sensitivity of the qPCR based on the amplification of Hsp70 remains to be determined in future studies. On the other hand, although xenodiagnosis showed low sensitivity, the utility of this method to evaluate infection transmission, evaluation of immune response or chemical treatment is still relevant. In future studies the sensitivity of xenodiagnosis could be increased either by feeding sand flies from other sites like ear (where parasites presence was demonstrated), evaluating infection in the sand flies by PCR techniques and using natural vectors. Finally, determining both the migration and the persistence of infection in various animal species, including humans, is a concluding factor to incriminate the disease reservoirs.
Acknowledgements:Authors are grateful to A. Daza and A. Restrepo VMD for their assistance in animal experimentation and L. López for her help with the statistical analysis. The advice of Dr. C. Muskus in molecular techniques is also appreciated. Also to K. Mondragon-Shem and A. Álvarez for assistance with language editing. Conflicts of interest: The manuscript was prepared and revised by all authors, who declare the absence of any conflict which can put the validity of the presented results at risk. Funding: CODI (project grant 20097000) and Group of Biogenesis sustainability of University of Antioquia.
BIBLIOGRAPHY
1. ALVAR, J.; VÉLEZ, I.D.; BERN, C.; HERRERO, M.; DESJEUX, P.; CANO, J.; JANNIN, J.; DEN BOER, M. 2012. WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS One. 7(5):e35671.
2. BARRAL, A.; GUERREIRO, J.; BOMFIM, G.; CORREIA, D.; BARRAL-NETTO, M.; CARVALHO, E.M. 1995. Lymphadenopathy as the first sign of human cutaneous infection by Leishmania braziliensis. Am. J. Trop. Med. Hyg. 53(3):256-9.
3. BOGDAN, C.; GESSNER, A.; SOLBACH, W.; ROLLINGHOF, M. 1996. Invasion, control and persistence of Leishmania parasites. Curr. Opin. Immunol. 8(4):517-25.
4. DAVOUST, B.; MARY, C.; MARIÉ, J.L. 2014. Detection of Leishmania in red foxes (Vulpes vulpes) from southeastern france using real-time quantitative PCR. J. Wildl. Dis. 50(1):130-2.
5. DE PAIVA, C.M.; DANTAS-TORRES, F.; DA CUNHA GONÇALVES DE ALBUQUERQUE, S.; SILVA DE MORAIS, R.C.; DE BRITO, M.E.; OTRANTO, D.; BRANDÃO-FILHO, S.P. 2013. Quantitative real time PCR assays for the detection of Leishmania (Viannia) braziliensis in animals and humans. Mol. Cell. Probes. 27(3-4):122-8.
6. DE RUITER, C.M.; VAN DER VEER, C.; LEEFLANG, M.M.; DEBORGGRAEVE, S.; LUCAS, C.; ADAMS, E.R. 2014. Molecular tools for diagnosis of visceral leishmaniasis: systematic review and meta-analy sis of diagnostic test accuracy. J. Clin. Microbiol. 9:3147-55.
7. FOLGUEIRA, C.; REQUENA, J.M. 2007. A postgenomic view of the heat shock proteins in kinetoplastids. FEMS Microbiol. Rev. 31(4):359-377.
8. FRAGA, J.; MONTALVO, A.M.; DE DONCKER, S.; DUJARDIN, J.C.; VAN DER AUWERA, G. 2010. Phylogeny of Leishmania species based on the heat-shock protein 70 gene. Infect. Genet. Evol. 10(2):238-245
9. GARCIA, L.; KINDT, A.; BERMUDEZ, H.; LLANOSCUENTAS, A.; DE DONCKER, S.; AREVALO, J.; WILBER, QUISPE.; TINTAYA, K.; DUJARDIN, J.C. 2004. Culture-independent species typing of neotropical Leishmania for clinical validation of a PCRbased assay targeting heat shock protein 70 genes. J. Clin. Microbiol. 42(5):2294-2297.
10. GARCIA, A.L.; PARRADO, R.; DE DONCKER, S.; BERMUDEZ, H.; DUJARDIN, J.C. 2007. American tegumentary leishmaniasis: direct species identification of Leishmania in non-invasive clinical samples. Trans. R. Soc. Trop. Med. Hyg. 101(4):368-371.
11. GONZÁLEZ, L.M.; VÉLEZ, I.D. 2006. Miltefosine for disseminated cutaneous leishmaniasis.Biomedica. 26(Suppl 1):13-16.
12. GRAÇA, G.C.; VOLPINI, A.C; ROMERO, G.A.; OLIVEIRA NETO, M.P.; HUEB, M.; PORROZZI, R.; BOITÉ, M.C.; CUPOLILLO, E. 2012. Development and validation of PCR-based assays for diagnosis of American cutaneous leishmaniasis and identification of the parasite species. Mem. Inst. Oswaldo Cruz. 107(5):664-674.
13. HOMMEL, M.; JAFFE, C.L.; TRAVI, B.; MILONI, G. 1995. Experimental models for leishmaniasis and for testing anti-leishmanial vaccines. Ann. Trop. Med. Parasitol. 89(Suppl 1):55-73.
14. MARTÍNEZ, J.E.; TRAVI, B.L,; VALENCIA, A.Z,; SARAVIA, N.G. 1991 Metastatic capability of Leishmania (Viannia) panamensis and Leishmania (Viannia) guyanensis in golden hamsters. J. Parasitol. 77(5):762-768.
15. MARTÍNEZ, J.E.; VALDERRAMA, L.; GAMA, V.; LEIBY, D.A.; SARAVIA, N.G. 2000. Clonal diversity in the expression and stability of the metastatic capability of Leishmania guyanensis in the golden hamster. J. Parasitol. 86(4):792-799.
16. MENDONÇA, M.G.; DE BRITO, M.E.; RODRIGUEZ, E.H.; BANDEIRA, V.; JARDIM, M.L.; ABATH, F.G. 2004. Persistence of Leishmania parasites in scars after clinical cure of american cutaneous Leishmaniasis: Is there a tterile cure? J. Infect. Dis. 189(6):1018- 1023.
17. MONTALVO, A.M.; MONZOTE, L.; FRAGA, J.; MONTANO, I.; MUSKUS, C.; MARIN, M.; DE DONCKER, S.; VÉLEZ, I.D.; DUJARDIN, J.C. 2008. PCR-RFLP and RAPD for typing neotropical Leishmania. Biomedica. 28(4):597-606.
18. MONTALVO, A.M.; NODARSE, J.F.; GOODRIDGE, I.M.; FIDALGO, L.M.; MARIN, M.; VAN DER AUWERA, G.; DUJARDIN, J.C.; VÉLEZ, I.D.; MUSKUS, C. 2010. Differentiation of Leishmania (Viannia) panamen- sis and Leishmania (V.) guyanensis using BccI for hsp70 PCR-RFLP. Trans. R. Soc. Trop. Med. Hyg. 104(5):364-367
19. NICOLAS, L.; SIDJANSKI, S.; COLLE, J.H.; MILON, G. 2000. Leishmania major reaches distant cutaneous sites where it persists transiently while persisting durably in the primary dermal site and its draining lymph node: a study with laboratory mice. Infect. Immunology. 68(12):6561-6566.
20. OSORIO, Y.; MELBY, P.C.; PIRMEZ, C.; CHANDRASEKAR, B.; GUARIN, N.; TRAVI, B.L. 2003. The site of cutaneous infection influences the immunological response and clinical outcome of hamsters infected with Leishmania panamensis. Parasite Immunol. 25(3):139-148.
21. ROBLEDO, S.M.; CARRILLO, L.M.; DAZA, A.; RESTREPO, A.M.; MUÑOZ, D.L.; TOBON, J.; MURILLO, J.D.; LOPEZ, A.; RIOS, C.; MESA, C.V.; UPEGUI, Y.A.; VALENCIA-TOBON, A.; MONDRAGON-SHEM, K.; RODRIGUEZ, B.; VÉLEZ, I.D. 2012. Cutaneous leishmaniasis in the dorsal skin of hamsters: a useful model for the screening of antileishmanial drugs. J. Vis. Exp. 21(62) pii: 3533.
22. ROMERO, I.; TELLEZ, J.; SUAREZ, Y.; CARDONA, M.; FIGUEROA, R.; ZELAZNY, A.; SARAVIA, N. 2010. Viability and burden of Leishmania in extralesional sites during human dermal leishmaniasis. PLoS Negl. Trop. Dis. 14:4(9). pii: e819.
23. ROQUE, A.L.R.; JASEN, A. 2014. Wild and synanthropic reservoirs of Leishmania species in the Americas. Int. J. Parasitol. Parasites Wild. 3(3):251-262
24. RUIJTER, J.M.; RAMAKERS, C.; HOOGAARS, W.M.; KARLEN, Y.; BAKKER, O.; VAN DEN HOFF, M.J.; MOORMAN, A.F. 2009. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37(6):e45.
25. SANTOS, T.R.; CARREIRA, V.S.; FERRARI, H.F.; MOREIRA, M.A.; LUVIZOTTO, M.C. 2014. Comparison of PCR with stained slides of bone marrow and lymph nodes aspirates with suspect diagnosis for leishmaniasis. Acta Trop. 140C:137-140
26. TITUS, R.G.; MARCHAND, M.; BOON, T.; LOUIS, J.A. 1985. A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunol. 7(5):545-555.
27. TOZ, S.O.; CULHA, G.; ZEYRECK, F.Y.; ERTABAKLAR, H.; ALKAN, M.Z.; VARDALI, A.T.; GUNDUZ, C.; OZBEL, Y. 2013. A real-time ITS1-PCR based method in the diagnosis and species identification of Leishmania parasite from human and dog clinical samples in Turkey. PLoS Negl. Trop. Dis. 7(5):e2205.
28. TRAVI, B.L.; OSORIO, Y.; MELBY, P.C.; CHANDRASEKAR, B.; ARTEAGA, L.; SARAVIA, N.G. 2002. Gender is a major determinant of the clinical evolution and immune response in hamsters infected with Leishmania spp. Infect. Immun. 70(5):2288-2296
29. VÉLEZ, I.D.; CARRILLO, L.M.; LÓPEZ, L.; RODRÍGUEZ, E.; ROBLEDO, S.M. 2012. An epidemic outbreak of canine cutaneous leishmaniasis in Colombia caused by Leishmania braziliensis and Leishmania panamensis. Am J Trop Med Hyg. 86(5):807-811.
30. VERGEL, C.; PALACIOS, R.; CADENA, H.; POSSO, C.J.; VALDERRAMA, L.; PEREZ, M.; WALKER, J.; TRAVI, B.L.; SARAVIA, N.G. 2006. Evidence for Leishmania (Viannia) parasites in the skin and blood of patients before and after treatment. J. Infect. Dis. 15;194(4):503-511
31. WALTON, B.C.; SHAW, J.J.; LAINSON, R. 1977 Observations on the in vitro cultivation of Leishmania braziliensis. J. Parasitol. 63(6):1118-1119.
Received: 22 January 2014 Accepted: 22 October 2014

Revista U.D.C.A Actualidad & Divulgación Científica por Universidad de Ciencias Aplicadas y Ambientales se distribuye bajo una Licencia Creative Commons Atribución-NoComercial 4.0 Internacional.